Существует масса наследственных заболеваний, без лечения которых люди живут недолго. Одно из таких – болезнь Марфана (код МКБ – Q87.4). Встречается редко, распространенность – 1 случай на 10000 человек. Риск рождения ребенка с данной аномалией возрастает, если заболевание Марфана есть у родителей. Узнайте, как проявляется арахнодактилия.
- Синдром Марфана – что это такое
- Тип наследования
- Синдром Марфана – причины
- Симптомы
- Диагностика
- Синдром Марфана – лечение
- Синдрома Марфана – продолжительность жизни
- Причины
- Классификация
- Симптоматика
- Скелет
- Зрительный аппарат
- Сердце и кровеносные сосуды
- Нервная система
- Кожный покров
- Лёгкие
- Диагностика
- Лечение
- Содержание
- Эпидемиология [ править | править код ]
- История [ править | править код ]
- Симптомы [ править | править код ]
- Лечение [ править | править код ]
Синдром Марфана – что это такое
Заболевание наследственное, возникает вследствие нарушения синтеза белка фибриллина. Формируется у плода во время внутриутробного развития, характеризуется изменениями скелета. Марфаноподобный синдром проявляется по-разному: чаще аномалия затрагивает глаза, сердечно-сосудистую систему, опорно-двигательный аппарат. Для диагностики используются различные методы. Все проявления патологии так или иначе связаны с повышенной растяжимостью тканей.
Тип наследования
Заболевание возможно у человека любой расы и любого пола. Тип наследования синдрома Марфана – аутосомно-доминантный. Мутация проявляется всегда, степень выраженности симптомов зависит от генетических особенностей. Из-за нарушения синтеза белка фибриллина соединительные ткани организма теряют прочность, что отражается на стенках сосудов, связочном аппарате. Лишь 25 % всех случаев аутосомного заболевания Марфана являются впервые возникшей мутацией в роду, где ранее синдром Марфана не встречался.
Синдром Марфана – причины
Генетическое заболевание очень редкое и недостаточно обследованное. Если говорить о причинах синдрома Марфана, самым правильным считается предположение о генетической мутации белка фибриллина. Это происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде. Можно выделить основные причины, способствующие появлению синдрома:
- наследственность;
- возраст отца (старше 35 лет).

Симптомы
Если у человека синдром паучьих пальцев, болезнь можно определить по его внешности. Человек отличается высоким ростом, непропорционально длинными конечностями, очень тонкими пальцами рук и ног. Кроме астенического телосложения характерны маленькая челюсть, нарушение прикуса, глубоко посаженные глаза, килевидная грудная клетка. У всех больных одинаковая форма черепа – удлиненная (долихоцефалия). Симптомы синдрома Марфана проявляются в зависимости от поражения отдельных органов и систем, составляют классическую триаду. Это могут быть:
- воронкообразная (вдавленная) грудная клетка;
- деформация позвоночника, сколиоз;
- обызвествление клапанного кольца митрального клапана;
- плоскостопие;
- гипермобильность;
- тахикардия;
- короткое туловище;
- помутнение, подвывихи, эктопия хрусталика;
- повышение глазного давления;
- близорукость;
- несимметричность зрачков;
- протрузия вертлужной впадины;
- проблемы с аортой (расширение, расслоение), которые могут вызвать летальный исход;
- нарушенная работа сердечно-сосудистой системы (фибрилляция предсердий, желудочная тахикардия);
- мышечная гипотония;
- инфекционный эндокардит;
- кифоз;
- вывихи шейного отдела;
- аннулоаортальная эктазия;
- прогнатия челюсти;
- ишемические, геморрагические инсульты;
- умственная одаренность;
- стеноз легочной артерии;
- коарктация аорты;
- поражение нервной системы;
- миксоматозная дегенерация створок митрального клапана;
- дилатация камер сердца;
- поражение митрального клапана сердца;
- несахарный диабет;
- выбросы адреналина;
- сочетанное поражение митрального клапана;
- ДМЖП, ДМПП;
- развитие неординарных способностей;
- разрыв створочных хорд;
- боль в суставах, костях;
- спондилолистез;
- аркообразное небо.
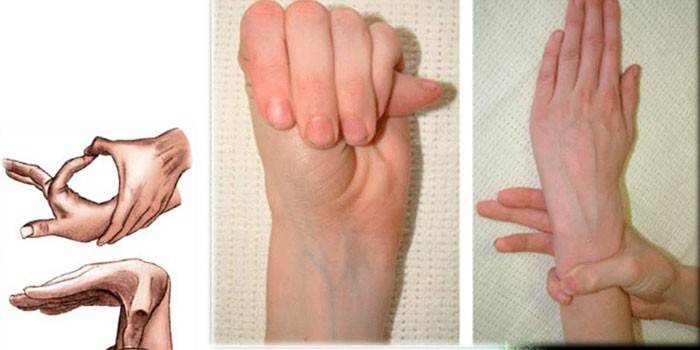
Диагностика
Для определения точного диагноза синдрома паучьих пальцев врачи собирают семейный анамнез, проводят внешний осмотр, обязательно назначают генетическое исследование, КГ, ЭхоКГ и так далее. Специалисты определяют соотношение роста человека и его кистей, выявляют деформацию грудной клетки, кифосколиоз, наличие долихостеномелии, проводят тесты Варги, охвата запястья и так далее.
Благодаря рентгенографии врачи обнаруживают увеличенные размеры левого желудочка, диагностируют протрузию вертлужной впадины, расширение дуги аорты. Для диагностики синдрома Марфана часто используют ЭхоКГ, на котором видны пролапс митрального клапана, дилатация аорты. Если есть подозрение на проблемы с аортой, назначается аортография, для диагностирования эктопии хрусталика – офтальмоскопию. При диагностике синдрома паучьих пальцев специалисты сравнивают болезнь с другими.
Синдром Марфана – лечение
Арахнодактилия не лечится. Симптомы, появляющиеся по мере прогрессирования долихостеномелии, устраняются разными способами. Главная цель медикаментозной терапии – не допустить развития осложнений. Если, к примеру, на первом году жизни у ребенка диагностировали марфаноподобный фенотип, аневризму аорты, сразу назначают средства для предотвращения ее прогрессирования. При диаметре аорты до 4 см выписывают ингибиторы АПФ, адреноблокаторы, при диаметре более 5 см, возможно хирургическое вмешательство. Больной должен постоянно наблюдаться у кардиолога.
Лечение синдрома Марфана заключается и в коррекции зрения. Больному подбирают правильный очки, в сложных случаях используют лазерный или хирургический методы. Если у ребенка marfan syndrome, наблюдаются скелетные нарушения, рекомендованы торакопластика, эндопротезирование. Курс лечения включает метаболическую терапию, прием витаминов, препаратов с глюкозамина сульфатами, янтарной кислоты, при слишком быстром росте – гормонов. Людям с марфаноидным фенотипом стертой формы показаны физические занятия.
Синдрома Марфана – продолжительность жизни
Если раньше люди с выраженной формой арахнодактилии жили недолго, то сейчас средняя продолжительность жизни составляет 40-50 лет. Это возможно при постоянном наблюдении у разных врачей, правильной профилактике, здоровом образе жизни. Продолжительность жизни при синдроме Марфана во многим зависит от того, проводится или нет хирургическая коррекция офтальмологических, суставных, сердечных нарушений. Комплексный подход, своевременное лечение долихостеномелии позволяют повысить качество жизни человека с диагнозом арахнодактилия даже при неонатальной форме.

- Боль в животе
- Высокий скелет
- Деформация грудной клетки
- Деформация пальцев
- Длинное лицо
- Изменение дислокации хрусталика
- Изогнутое небо
- Искривление позвоночника
- Непропорциональные конечности
- Одышка
- Онемение конечностей
- Плоскостопие
- Повышенная утомляемость
- Появление растяжек
- Слабость в ногах
- Тонкий скелет
- Узкое лицо
- Учащенное сердцебиение
Синдром Марфана – наследственная патология, в результате прогрессирования которой происходит поражение соединительной ткани. В процесс также вовлекается скелетно-мышечная система и зрительный аппарат. Ткань внутренней среды (соединительная) выполняет массу важных функций, одной из которых является соединение определённых частей тела, и закладывание основы для их нормального роста и развития. В случае прогрессирования синдрома Марфана, в структуре ткани появляются дефекты, которые не дают ей функционировать так, как необходимо.
Так как данная ткань располагается по всему телу, синдром Марфана оказывает влияние и на остальные органы и системы:
Синдром Марфана в одинаковой мере поражает и мужчин, и женщин. Патологию диагностируют у людей всех этнических групп и рас. Распространённость – 1 случай на 10 тысяч человек. Риск рождения ребёнка с данной болезнью повышается в несколько раз в том случае, если отец достигает возраста 35 лет. В 50% случаев ребёнок рождается с патологией, если у одного из родителей диагностирован синдром Марфана. Тип наследования – аутосомно-доминантный.
Причины
Основная причина прогрессирования синдрома Марфана – мутация гена FBN1. Он отвечает за синтез фибриллина. Данный белок выполняет чрезвычайно важную функцию в организме – он отвечает за сократимость и эластичность соединительной ткани.
Нарушение формирования волокон соединительной ткани и утрата ими прочности, происходит ещё во время внутриутробного развития плода. Это приводит к тому, что ткань перестаёт выдерживать естественные нагрузки. Атипичные изменения наблюдаются в крупных сосудах мышцах, клапанах сердца. Если не проводить адекватную терапию, то продолжительность жизни человека, больного синдромом Марфана, составляет всего 40 лет. Правильно подобранный план лечения даёт возможность увеличить эту цифру в два раза.
Классификация
В медицине выделяют несколько форм синдрома Марфана:
Классификация по тяжести протекания болезни:
В зависимости от характера течения, недуг может быть:
Симптоматика

Синдром Марфана может проявляться у разных людей по-разному. У некоторых признаки данного типа патологии выражены очень ярко, у других же наблюдаются исключительно лёгкие симптомы. Обычно прогрессирование симптомов происходит по мере того, как человек растёт.
Признаки болезни зависят от того, какие системы и органы в организме поражены, поэтому симптоматика может несколько изменяться.
Скелет
- пациенты с данным типом патологии имеют гибкий, высокий и тонкий скелет. Это обусловлено тем, что при прогрессировании патологии в первую очередь поражаются длинные кости скелета;
- конечности, пальцы рук и ног часто являются непропорционально длинными относительно остальных частей тела человека;
- лицо длинное и узкое;
- изменение формы неба – оно становится изогнутым. Такая деформация приводит к нарушению линии зубов;
- плоскостопие;
- выступающая или вдавленная грудина;
- сколиоз.
Зрительный аппарат
Признаки нарушения работы зрительного аппарата наблюдаются более чем у половины больных синдромом Марфана. Наиболее распространённый симптом – изменение дислокации хрусталика. При осмотре глаза можно отметить, что хрусталик смешен выше или ниже своего нормального положения. Смещение может быть как минимальным, так и ярко выраженным. На фоне данной болезни может также развиться серьёзное осложнение – отслоение сетчатки.
Некоторые пациенты с данным диагнозом страдают близорукостью, ранней глаукомой или катарактой. Симптомы могут проявиться ещё в детском возрасте и по мере взросления человека они только усиливаются.
Сердце и кровеносные сосуды
- потеря эластичности стенок аорты. Данное состояние именуют расширением аорты. Это состояние крайне опасное, так как существует высокий риск того, что она разорвётся и это приведёт к смерти пациента;
- дефекты клапанов также являются опасным состоянием. Они могут «протекать», создавая при этом шумы в сердце. Если дефект в клапане небольшой, то никаких симптомов может не возникнуть. Но большие дефекты становятся причиной появления учащённого сердцебиения, одышки, повышенной утомляемости.
Нервная система
Спинной и головной мозг окружены жидкостью, которая локализуется в мембране. В медицине её именуют твердой мозговой оболочкой, и состоит она из соединительной ткани. По мере того как пациент с данным типом наследственной патологии становится старше, оболочка ослабевает. В результате этого увеличивается давление на позвонки в нижней части позвоночного столба. Клиницисты именуют такое состояние дуральной эктазией.
Такие патологические изменения могут вызывать лишь чувство лёгкого дискомфорта, а могут стать причиной появления сильных болей в животе, слабости в ногах, онемения конечностей.
Кожный покров
У людей с синдромом Марфана обычно на коже появляются растяжки без видимых на то причин (изменение массы тела). Они могут проявиться в любом возрасте и, как правило, дискомфорта не доставляют. Но стоит отметить, что люди с данной болезнью подвергаются повышенному риску образования паховой или брюшной грыжи.
Лёгкие
Признаки поражения лёгких у пациентов с этой болезнью выражены не ярко. Дефекты внутренней ткани делают воздушные мешочки в лёгких менее эластичными. Однако стоит учесть, что если по какой-либо причине они опухнут и начнут растягиваться, может возрасти риск развития рака лёгких.
Такие признаки нарушения дыхания во сне, как храп или апноэ, у людей с синдромом Марфана проявляются редко.
Диагностика
Диагностика синдрома Марфана основывается на анализе семейного анамнеза пациента. Тщательно изучается физическое и нервно-психическое состояние человека. Для точной постановки диагноза требуется наличие хоть одного из указанных симптомов:
- изменение дислокации хрусталика;
- деформация грудной клетки;
- кифосколиоз;
- арахнодактилия;
- аневризма аорты.

Также должны присутствовать ещё два признака, указывающих на болезнь:

Обычно диагностика болезни не составляет особого труда. Но иногда врачам требуется провести дополнительные рентген-функциональные методики исследования. Диагностикой синдрома Марфана занимаются следующие специалисты:
Лечение
Лечение синдрома Марфана необходимо проводить только после проведения диагностики. Причина в том, что симптомы патологии могут указывать и на другие заболевания. При помощи дифференциальной диагностики их можно исключить.
Специфического лечения синдрома Марфана не разработано, так как нет никакого способа изменить гены ребёнка ещё до его рождения. Поэтому проводится симптоматическое лечение изменений, которые происходят в организме из-за болезни. Некоторые можно устранить консервативными методиками, для устранения других иногда требуется проведение оперативного лечения.
Пациент с синдромом Марфана наблюдается сразу у нескольких специалистов – офтальмолога, кардиолога, невролога и прочее. Основное направление терапии – поддержание нормального функционирования сосудов и сердца для сохранения жизни пациента.
Методики лечения болезни:
- приём лекарственных препаратов. В терапию включают антикоагулянты, адреноблокаторы, антиаритмические препараты и прочее;
- хирургическая операция показана в случае наличия пороков сердца, аорты. Возможно проведение протезирования клапанного аппарата;
- нормализация зрения проводится за счёт коррекции миопии, устранения катаракты и глаукомы. Иногда требуется провести имплантацию искусственного хрусталика;
- тракция (при сколиозе);
- операция проводится при поражении позвоночного столба и суставов (пластика суставов, удаление межпозвоночных грыж, протезирование);
- физиолечение и занятие ЛФК;
- если болезнь поразила лёгкие, то проводится их дренирование.
| Синдром Марфана | |
|---|---|
 |
|
| МКБ-10 | Q 87.4 87.4 |
| МКБ-10-КМ | Q87.4 и Q87.40 |
| МКБ-9 | 759.82 759.82 |
| МКБ-9-КМ | 759.82 [1] |
| OMIM | 154700 |
| DiseasesDB | 7845 |
| MedlinePlus | 000418 |
| eMedicine | ped/1372 orthoped/414 orthoped/414 |
| MeSH | D008382 |
Синдром (болезнь) Марфана — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутацией гена, кодирующего синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами [2] , и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Содержание
Эпидемиология [ править | править код ]
Синдром Марфана — редкое заболевание с классическим менделевским наследованием. Распространённость в популяции составляет порядка 1 на 5000. Синдром диагностируется во всем мире, в любых этнических группах.
История [ править | править код ]
Впервые признаки заболевания были описаны в 1875 году американским офтальмологом Э. Вильямсом (англ. E. Williams ), описавшим эктопию хрусталика у брата и сестры, которые были исключительно высокими и имели гипермобильные суставы от рождения [3] . В последующие годы эта болезнь наблюдалась французским профессором педиатрии Антуаном Марфаном [fr] , который представил в 1896 году клиническое наблюдение 5-летней девочки Габриэль с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя. [4]
Позднее выяснилось, что в действительности девочка страдала врождённой контрактурной арахнодактилией. [5]
Американский генетик Виктор Маккьюсик открыл этим синдромом новую нозологическую страницу наследственных заболеваний соединительной ткани. [6]
Симптомы [ править | править код ]
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами. [7]
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).
Диагностика синдрома Марфана (СМ) сегодня основана на Гентских критериях (DeРаере A. et al.,1996) и пересмотра Гентских критериев в 2010 году (J.Med.Genet.2010; 476-485). В основу алгоритма диагностики положено выделение больших и малых критериев, характеризующих выраженность изменений соединительной ткани в различных органах и системах.
Лечение [ править | править код ]
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм. [8]







