Талассемия – это патология крови, для которой характерны нарушения в формировании одной или сразу нескольких цепей гемоглобина в клетках эритроцитов. Это становится причиной развития малых, некачественных эритроцитов.
При таком заболевании наблюдается ухудшение транспортировки кислорода к тканям и клеточным элементам организма. Это вызывает их гипоксию, становятся очевидными проявления признаков болезни.
Раньше недуг чаще наблюдался в климатически жарких местах, где люди страдают малярией: Юго-Восточной Азии, Африке, странах Средиземноморья. Но в современном мире он диагностируется в самых разных регионах из-за миграции населения. Патология формируется одинаково у лиц женского и мужского пола.
.jpg)
Ежегодно рождается около 200 000 детей с этой болезнью, у 50% из них диагностируется гомозиготная форма β-талассемии. Последняя быстро приводит к смерти.
При гетерозиготной форме болезнь себя не проявляет, это возможно только в экстремальных ситуациях. Однако высок риск ее развития у потомков, они могут получить ее как наследственную патологию.
- Причины талассемии
- Классификация
- Симптомы и проявления
- Признаки большой талассемии
- Симптомы малой талассемии
- Диагностика заболевания
- Лечение
- Возможные осложнения
- Профилактика
- Прогнозы
- Что такое талассемия?
- Классификация талассемии
- Альфа-талассемия
- Бета-талассемия
- Какие обследования, анализы нужны?
- Лечение талассемии
- Видеозаписи по теме
- Что представляет собой бета-талассемия
- Почему происходит разрушение эритроцитов
- Разновидности бета-талассемии
- Малая форма бета-талассемии
- Большая бета-талассемия
- Диагностирование заболевания
- Терапия заболевания
- Лечение посредством гемотрансфузии
- Лечение народными рецептами
- Прогноз заболевания
- Профилактические меры
Причины талассемии
Они изучены хорошо, поскольку заболевание достаточно распространено. Достоверно известно, что первые изменения наблюдаются при нарушении синтеза белковых цепей гемоглобина, который способен связываться с молекулами кислорода и транспортировать их ко всем тканям организма.
Чаще всего свое развитие заболевание получает из-за генов, которые дети наследуют от своих родителей. В тех из них, которые несут ответственность за синтез гемоглобина, обнаруживается мутация.
При изучении структуры обнаруживается пигмент, в состав которого входит железо и 2 пары соединений белка: альфа- и бета-цепи. Если синтез одной из них нарушается, то накапливается вторая.

Провоцируют такие изменения те гены, что несут ответственность за формирование белка гемоглобина. При такой мутации идет нарушение правильного набора аминокислот в цепочке. Явление характерно и для возбудителя малярии.
Наследственность у ребенка способна проявиться в двух вариантах:
- Гомозиготный вид патологии – от мамы и папы одновременно.
- Гетерозиготный – от одного из родителей.
Классификация
Если аномальный белок гемоглобина наследуется с обеих родительских сторон, то это тяжелая форма, для которой характерны ярко выраженные проявления. Это большая талассемия, или болезнь Кули (в честь доктора, что ее описал).
.jpg)
Она наследуется по типу рецессивному (двухаллельная система), в основе лежит снижение синтеза цепей полипептидов.Если это идет со стороны одного родителя, то при отсутствии крайне неблагоприятных обстоятельств признаки отсутствуют.
В современной классификации выделены следующие формы недуга:
- Альфа-талассемия. Наблюдается при отсутствии одного либо нескольких генов, которых в норме должно быть по два от матери и отца. Именно такое количество обеспечивает достаточную выработку белков цепочки альфа-глобина. При отсутствии двух генов возможно проявление симптомов анемии. При недостатке трех генов диагностируется гемоглобинопатия H, которая ярко выраженно проявляется симптомами малокровия. Отсутствие четырех генов – явление довольно редкое, которое носит название “водянка плода”. Такие дети умирают до, во время либо сразу после родов.
- Бета-талассемия наблюдается при нарушении одного либо двух генов, которых в норме идет по одному от папы и мамы. Вырабатывается недостаточное количество белка бета-глобина. При видоизменении одного гена человек является носителем болезни, не исключено проявление умеренного малокровия. При изменении обоих генов одновременно диагностируется бета-талассемия интермедия (анемия Кули). При основной развивается и тяжелая форма анемического синдрома. Гомозиготная бета-форма выявляет себя к году жизни ребенка и различается проявлением признаков. Гетерозиготная (малая форма талассемии) характеризуется размытыми симптомами, либо признаки полностью отсутствуют.
Нужно учитывать, что в отдельных случаях гомозиготная форма протекает легко, ее нельзя квалифицировать как большую талассемию, поэтому ее отмечают как промежуточную.
У этой патологии есть три степени:
- Тяжелая. Аномальные изменения настолько сильны, что ребенок умирает на первом году жизни.
- Среднетяжелая. Ребенок способен прожить около восьми лет.
- Легкая форма. Взрослый человек умирает, не дожив до старости.
Несмотря на общий диагноз, β-талассемия у людей может значительно различаться результатами исследований, проявлениями признаков, клиническим течением, лечением и прогнозами на будущее.
Симптомы и проявления
Признаки зависят от формы и времени появления мутации. Большая талассемия случается при гомозиготном происхождении, проявляется у детей сразу после рождения:
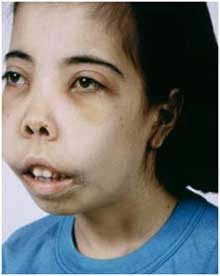
- Удлиненная кверху черепная коробка.
- Верхняя челюсть больше развита по сравнению с нижней.
- Лицевой скелет черепа напоминает монголоидный.
К году жизни наблюдаются следующие признаки:
- Расширение перегородки носа, приплюснутая его форма.
- Формирование аномальных костных наростов в области стоп.
- Нарушения прикуса.
- Желтушность кожных покровов из-за нарушения функции селезенки.
Дефицит кислорода в тканях вызывает такие симптомы:
- Увеличение печени, развитие раннего цирроза.
- Формирование сердечной недостаточности. Излишек железа скапливается на мышце миокарда и нарушает сократительную деятельность мышцы сердца.
- Запаздывание в физиологическом и интеллектуальном развитии.
- Формирование сахарного диабета.
У взрослых людей проявляются следующие симптомы:
- Формирование язв на кожных покровах, их провоцируют аномальные нарушения в кровообращении.
- Частые пневмонии.
- Регулярные переломы костей.
- Воспалительные процессы в желчном пузыре, связанные с отложением камней.
- Кардиосклероз — патология сердца, характеризующаяся разрастанием соединительного рубца в миокарде, замещением мышц и деформацией клапанов.
- Формирование сепсиса при инфекционных патологиях.
- Нарушения на половом уровне.
При развитии промежуточной формы заболевания не наблюдается изменений внешнего вида человека, умственное и физическое развитие в норме, однако есть увеличение селезенки и ломкость костей.
Признаки большой талассемии
Наблюдается сразу после появления на свет:
- Удлиненная форма черепа.
- Монголоидный тип лица.
- Увеличение верхней челюсти.
- Увеличение перегородки носа.
Проведение анализа крови указывает на гепатомегалию, которая заканчивается формированием цирроза и диабета.
.jpg)
Если при рождении ставится диагноз “талассемия”, причем признаки яркие, то чаще всего такие дети могут прожить около года.
Симптомы малой талассемии
Если генная мутация передалась от одного из родителей, то развивается малая талассемия. Симптомы в таком случае размытые либо не проявляются вообще. Клиническая картина следующая:
- Повышенная утомляемость.
- Ухудшение работоспособности.
- Регулярные боли в голове, беспричинные головокружения.
- Бледность кожи с проявлением желтушности.
- Увеличение селезенки.
Наибольшая опасность такого состояния заключается в повышении риска попадания инфекции в организм.
Диагностика заболевания
Обследование включает в себя несколько этапов:
- Сбор анамнеза. Определяются опасные патологии, которые диагностировались у женщины при беременности и могли стать причиной развития патологии.
- Оценка протекания периода вынашивания ребенка.
- Тщательный осмотр врачом: оценивается общее состояние пациента, состояние и цвет кожи, проводится пальпация брюшной области для определения гепатоспленомегалии.
- Опрос родителей либо самого пациента для выяснения выраженности симптомов и тяжести заболевания.
Также назначается проведение следующих лабораторных исследований:
- Биохимический анализ крови.
- ПЦР-тесты.
- Исследование биологического синтеза цепочек гемоглобина.
- Общее исследование крови.
- Электрофорез гемоглобина.
Также необходимо проведение инструментальных методов диагностики:
- УЗИ брюшной полости (обнаруживает увеличение селезенки).
- Рентген костей.
- Пункция костного мозга.
- Развитие плода в утробе матери.
При обнаружении гомозиготной формы патологии назначается искусственное прерывание беременности.
Лечение
Определив, что это такое и каковы признаки, необходимо выбрать методы терапии. Лечебные меры в первую очередь направляют на нормализацию количества гемоглобина.
Помимо этого, есть и другие методы лечения:
.jpg)
- При тяжелых формах недуга необходимо проводить частое переливание крови, но это дает временный результат.
- Назначается хелат железа.
- В современной медицине есть возможность проводить переливание размороженных либо отфильтрованных эритроцитов. Такая процедура более безопасна для детей.
- При увеличенной селезенке назначается ее удаление. Хирургическое лечение возможно только после 5 лет жизни, необходимо учитывать возрастание риска проникновения инфекции.
- Трансплантация костного мозга, но найти донора проблематично.
К клиническим рекомендациям относится специальная диета, которая подразумевает прием орехов, чая, сои, какао (продуктов, которые противостоят усвоению железа). Прием аскорбиновой кислоты, которая способствует выводу железа из организма.
Возможные осложнения
При умеренных и тяжелых формах лечебные меры направлены на продление периода жизни и устранение возможных осложнений, таких как:
- Патологии сердца и печени. Высокий уровень железа в организме может стать причиной повреждения различных тканей, больше всего этому подвержены сердечная мышца и печень. Это нередко становится причиной смертельного исхода, поэтому необходимо проведение регулярного переливания крови.
- Проникновение инфекции в организм также нередко является причиной смерти пациента, особенно это касается тех, у кого проводилась операция по удалению селезенки.
- Остеопороз. При такой патологии костные элементы подвержены частым переломам.
Профилактика
Вылечить талассемию невозможно, поэтому необходимо принимать меры профилактики, которые заключаются в следующем:
.jpg)
- Осуществление диагностики во время беременности, особенно если у обоих родителей наблюдается талассемия.
- Проведение генетического исследования.
- При необходимо проводится искусственное прерывание беременности.
Прогнозы
При диагностировании малой талассемии прогнозы хорошие, человек ведет нормальную жизнь, продолжительность которой практически как у здорового.
При бета-талассемии лишь некоторые больные способны дожить до половозрелого возраста.
Тяжелая гомозиготная форма требует постоянного лечения, поскольку только такие мероприятия, как переливание крови, поддерживают жизнь пациента.
Что такое талассемия?
Талассемия (анемия Кули) — это группа наследственных расстройств, которые влияют на количество гемоглобина, производимых человеком. Гемоглобин относится к семейству соединений, состоящих из гема (железосодержащий комплекс) и различных глобинов (белковые цепи, которые окружают гемовый комплекс).
Молекулы гемоглобина (Hb) находятся во всех эритроцитах и являются причиной их красного цвета. Они связывают кислород в легких, переносят его через кровоток и выделяют в ткани организма. Различные типы гемоглобина классифицируются в соответствии с типом белковых цепей, которые они содержат.
Нормальные гемоглобины взрослых включают в себя:
- Гемоглобин А (составляет около 95% – 98% взрослого Hb). HbA содержит две альфа (α) белковые цепи и две бета (ß) цепи.
- HbA2 (составляет около 2% – 3,5% взрослого Hb), имеет две альфа (α) и две дельта (δ) цепи.
- HbF (до 2%). Это основной гемоглобин, вырабатываемый плодом во время беременности. Его производство обычно падает до низкого уровня в течение года после рождения. HbF имеет две альфа (α) и две гамма (γ) цепи.
Мутации в генах, кодирующих глобиновые цепи, могут вызывать нарушения в выработке гемоглобина. Есть 4 гена, которые кодируют цепи альфа-глобина, и 2 гена, которые кодируют цепи бета-глобина.
Наследственные нарушения производства гемоглобина делятся на две категории:
- Талассемия: снижение выработки нормальных гемоглобинов
- Гемоглобинопатия: производство аномальной молекулы гемоглобина
Талассемии представляют собой группу нарушений, при которых мутации в одном или нескольких из генов альфа- или бета-глобина вызывают уменьшение количества продуцируемого HbA. Это приводит к снижению уровня HbA, относительному увеличению количества минорных гемоглобинов HbA2 и HbF и, возможно, обнаружению необычных типов гемоглобина.
Талассемии обычно классифицируются по типу глобиновой цепи, синтез которой снижается.
Классификация талассемии
Альфа-талассемия
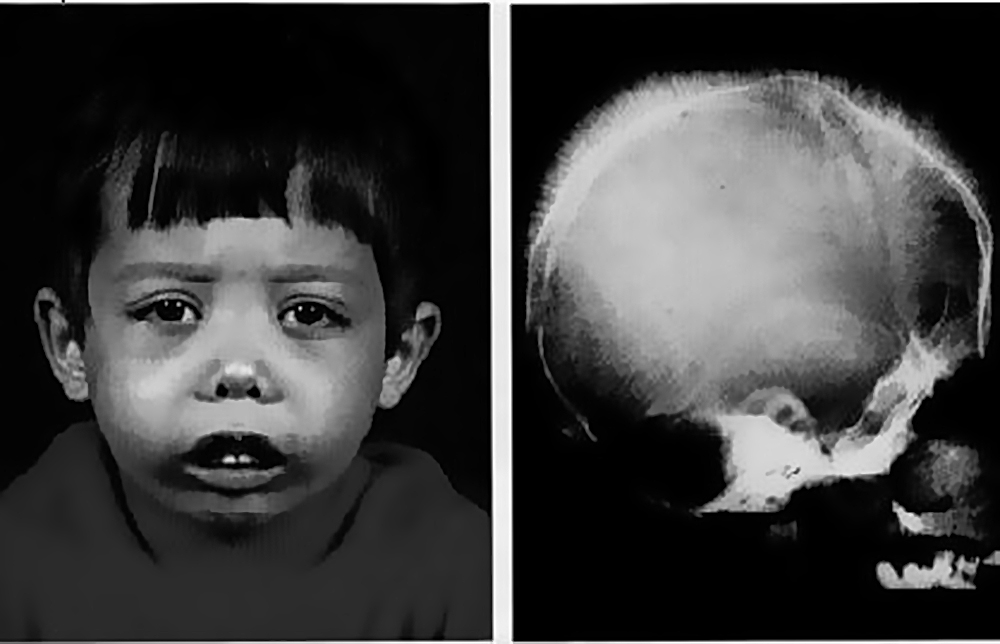
Альфа-талассемия возникает вследствие делеции или мутации в одной, или нескольких копиях гена 4-альфа-глобина. Чем больше затронутых генов, тем меньше вырабатывается альфа-глобина. Четыре различных типа альфа-талассемии включают в себя:
- Черта альфа-талассемии (1 пораженный ген). Безмолвный носитель будет иметь нормальные уровни гемоглобина и индексы эритроцитов, которые являются нормальными или демонстрируют слегка уменьшенный MCH (гипохромия). Носители могут передать пораженный ген своим потомкам. Часто эти люди идентифицируются только после рождения ребенка с болезнью HbH или признаком альфа-талассемии.
- Черта альфа-талассемии (2 пораженных гена). Пациенты с признаком альфа-талассемии имеют меньшие (микроцитарную), бледные (гипохромную) эритроциты и легкую хроническую анемию, но обычно они не испытывают никаких симптомов. Это анемия, которая не реагирует на добавки железа. Диагностика признака альфа-талассемии обычно происходит путем исключения других причин микроцитарной анемии. Подтверждающее тестирование с помощью анализа ДНК является важной частью процесса диагностики альфа-талассемии, поскольку на каждой хромосоме имеется две копии (HbA1 и HbA2) гена альфа-цепи. Если человек потерял обе копии из одной хромосомы, это имеет различные последствия для передачи более серьезной формы заболевания своим детям, чем если бы они потеряли одну копию из каждой двух разных хромосом. Смотрите раздел об анализе ДНК ниже.
- Болезнь гемоглобина Н (3 пораженных гена). При этом условии большое уменьшение количества производимых цепей альфа-глобина вызывает избыток бета-цепей, которые затем агрегируют в тетрамеры бета4 (группы из 4 бета-цепей), известные как гемоглобин H. Болезнь HbH может вызывать анемию от средней до тяжелой степени и спленомегалию (увеличенная селезенка). Клиническая картина, связанная с заболеванием HbH, чрезвычайно разнообразна. У некоторых людей она протекает бессимптомно, в то время как другие имеют тяжелую анемию. Болезнь гемоглобина Н чаще всего встречается у лиц юго-восточного или восточно-средиземноморского происхождения.
- Большая альфа-талассемия (также называемая фетальный гидропс, 4 пораженных гена). Это самая тяжелая форма альфа-талассемии. В этих условиях альфа-глобин не вырабатывается, следовательно, HbA или HbF также не вырабатываются. Плоды, пораженные альфа-талассемией, становятся анемичными на ранних сроках беременности. Они становятся отечными (сохраняют жидкость), и часто имеют увеличенные сердца и печень. Этот диагноз часто ставится в последние месяцы беременности, когда УЗИ плода указывает на отек плода. Приблизительно в 80% случаев у матери возникает токсемия и может развиться тяжелое послеродовое кровотечение. Дети с большой альфа-талассемией обычно недоношенные, мертворожденные или умирают вскоре после рождения.
Люди с альфа-талассемией могут быть неправильно диагностированы неосторожными врачами как дефицит железа, так как дефицит железа также приводит к появлению небольших бледных (микроцитарных гипохромных) эритроцитов.
Важно, чтобы лечение железом у больных талассемией назначалось только тогда, когда специфические анализы на железо (ферритин, сывороточное железо, TIBC, трансферрин) подтвердили дефицит железа. Это особенно важно при альфа-талассемии, где существует небольшая вероятность развития опасной перегрузки железом.
Альфа-талассемия чаще всего встречается у людей с этническим происхождением из Юго-Восточной Азии, Южного Китая, Ближнего Востока, Индии, Африки и Средиземноморья.
Бета-талассемия
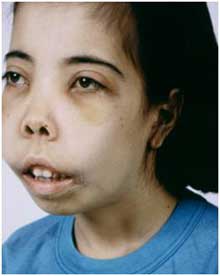
Бета-талассемия происходит из-за мутаций в одном или обоих генах бета-глобина. Было выявлено от 100 до 200 мутаций, но распространены только около 20.
Тяжесть анемии, вызванной бета-талассемией, зависит от того, какие мутации присутствуют, и от того, уменьшают ли они выработку бета-глобина (так называемая бета + талассемия) или полностью ее устраняют (так называемая бета-талассемия). Различные типы бета-талассемии следующие:
- Бета-талассемия с характерной чертой. У человека с этим заболеванием один нормальный ген и один с мутацией. Обычно они не испытывают никаких проблем со здоровьем, кроме микроцитоза (маленьких эритроцитов) и возможной легкой анемии, которая не будет реагировать на добавки железа. Эта генная мутация может передаваться детям индивидуума.
- Талассемия интермедиа. В этом состоянии у больного человека есть два ненормальных гена, но он все еще производит некоторое количество бета-глобина. Тяжесть возникшей анемии и проблемы со здоровьем зависят от имеющихся мутаций. Разграничительной линией между промежуточной талассемией и большой талассемией является степень анемии, а также количество и частота переливаний крови, необходимых для ее лечения. Тем, у кого промежуточная талассемия, иногда требуется переливание, но не на регулярной основе.
- Талассемия крупная. Это самая тяжелая форма бета-талассемии. У пациента есть два ненормальных гена, которые вызывают либо серьезное снижение, либо полное отсутствие продукции бета-глобина, предотвращая выработку значительных количеств HbA. Это состояние обычно появляется у ребенка после 3-месячного возраста и вызывает опасную для жизни анемию. Эта анемия требует регулярных переливаний крови в течение всей жизни и значительной постоянной медицинской помощи. Со временем эти частые переливания приводят к чрезмерному количеству железа в организме. При отсутствии лечения это избыточное железо может откладываться в печени, сердце и других органах и может привести к преждевременной смерти от недостаточности органов.
Другие формы талассемии возникают, когда ген бета-талассемии наследуется в сочетании с геном варианта гемоглобина. Наиболее важными из них являются:
- HbE – бета-талассемия. HbE является одним из наиболее распространенных вариантов гемоглобина, встречающихся преимущественно у людей юго-восточного азиатского происхождения. Если человек наследует один ген HbE и один ген бета-талассемии, комбинация провоцирует талассемию HbE-бета, которая вызывает умеренно тяжелую анемию, подобную промежуточной бета-талассемии.
- HbS – бета-талассемия или серповидноклеточная – бета-талассемия. HbS является одним из наиболее известных вариантов гемоглобина. Наследование одного гена HbS и одного гена бета-талассемии приводит к талассемии HbS-бета. Тяжесть состояния зависит от количества бета-глобина, продуцируемого бета-геном. Если бета-глобин не вырабатывается, клиническая картина практически идентична серповидноклеточной анемии.
Какие обследования, анализы нужны?
Общий клинический анализ крови (ОАК). ОАК — это анализ клеток и жидкости в крови. Помимо прочего, ОАК сообщит врачу, сколько эритроцитов в крови, сколько гемоглобина содержится в них, и поможет измерить размер и форму присутствующих эритроцитов. Эти переменные называются индексами эритроцитов и включают MCH (средний корпускулярный гемоглобин) и MCV (средний корпускулярный объем), как измерения содержания гемоглобина и размера эритроцитов.
Низкий MCH или MCV часто являются первым признаком талассемии. Если уровень MCH или MCV низкий, а дефицит железа исключен, человек может быть носителем талассемии. При дефиците железа трудно исключить талассемию.
Микроскопическое исследование мазка крови. При этом тесте тонкий микроскопический слой крови исследуют на предметном стекле под микроскопом. Количество и тип лейкоцитов, эритроцитов и тромбоцитов можно подсчитать вручную и оценить, являются ли они нормальными и зрелыми. Различные нарушения влияют на нормальное производство клеток крови. При талассемии эритроциты часто бывают микроцитарными (маленькими) с низким MCV. Они также могут:
- Быть гипохромным (бледный — указывает низкий гемоглобин, чем обычно).
- Варьироваться по размеру (анизоцитоз) и форме (пойкилоцитоз).
- Быть зародышем (не нормально в зрелом эритроците).
- Быть искаженными, выглядеть под микроскопом как яблочки.
Анализ железа. Анализ может включать: железо, ферритин, UIBC, TIBC и процентное насыщение трансферрина. Эти тесты измеряют различные аспекты хранения и использования железа в организме. Они помогают определить, является ли дефицит железа причиной и/или обострением анемии пациента. Одному или нескольким пациентам с талассемией также может быть назначено наблюдение за степенью перегрузки железом.
Оценка гемоглобинопатии (Hb). Этот тест измеряет тип и относительное количество гемоглобинов, присутствующих в эритроцитах. Гемоглобин А, состоящий из альфа- и бета-глобина, является основным нормальным типом гемоглобина, обнаруживаемым у взрослых. Больший процент HbA2 и/или HbF обычно наблюдается при признаке бета-талассемии. HbH может наблюдаться при альфа-талассемии, но только тогда, когда по меньшей мере два из четырех альфа-генов удалены или мутированы.
Анализ ДНК, Во многих случаях анализ ДНК не требуется, поскольку диагноз может быть поставлен по результатам вышеуказанных тестов. Анализ ДНК чаще всего используется в семьях, страдающих альфа-талассемией. Этот тест используется для изучения делеций и мутаций в альфа и реже генах, продуцирующих бета глобин.
Семейные исследования могут быть проведены, чтобы оценить статус носителя и типы мутаций, присутствующих в других членах семьи. Исследование ДНК является важным инструментом для установления точного диагноза талассемии. В частности, при альфа-талассемии важно знать, имеет ли человек с чертой альфа-талассемии два мутированных гена в одной хромосоме или по одному в каждой хромосоме.
Лечение талассемии
Большинство людей с талассемией не нуждаются в лечении. Всем людям с диагнозом талассемия, которые планируют семью, настоятельно рекомендуется обратиться за генетическим советом, чтобы понять последствия для своего потомства. Требуется, чтобы лаборатория провела генетическое исследование партнера, чтобы генетический консультант предоставил точную информацию о риске передачи затронутых генов своим детям и серьезности талассаземии или гемоглобинопатии, которая может развиться.
Пациенты с гемоглобином Н или промежуточной бета-талассемией будут испытывать различные степени анемии в течение всей жизни. Они могут вести относительно нормальную жизнь, но требуют регулярного мониторинга и иногда могут нуждаться в переливании крови.
Добавки с фолиевой кислотой часто назначаются для борьбы с анемией, но следует избегать приема препаратов железа, если дефицит железа не подтвержден более конкретными исследованиями.
Часто изменения в показателях эритроцитов наблюдаются, когда женщина беременна, это может иногда приводить к тяжелой анемии. Понимание влияния диагноза талассемии на беременность и возможных последствий для здоровья потомства пары является одной из наиболее важных причин для установления точного диагноза талассемии.
Больным с бета-талассемией обычно требуется переливание крови каждые 3-4 недели в течение всей жизни. Эти переливания помогают поддерживать гемоглобин в достаточно высокой концентрации, чтобы обеспечить организм кислородом и предотвратить нарушения роста и повреждения органов.
Частые переливания, однако, поднимают железо до токсичных уровней, что приводит к отложению железа в печени, сердце и других органах.
Регулярная хелатная терапия железом используется для снижения уровня железа в организме Это включает введение препарата, который связывается с железом и помогает вымывать его из организма через мочу. Также часто больным может требоваться спленэктомия (хирургическая операция по удалению селезёнки).
Дети с большой альфа-талассемией обычно рождаются недоношенными, мертворожденные или умирают вскоре после рождения. Лечение сосредоточено на выявлении состояния и прерывании беременности или наблюдении за осложнениями у матери.
Экспериментальные методы лечения, такие как переливание крови плода, были успешными в очень немногих случаях при рождении ребенка.
Видеозаписи по теме
Талассемия принадлежит категории заболеваний генетической этиологии. Процесс протекания патологии основан на том, что опасности подвергаются эритроциты – весомые кровяные элементы. Бета-талассемия представляет собой состояние, когда гемоглобином вырабатывается недостаточный объем бета-белков.
Среди лиц славянской группы недуг почти не распространен, ему подвержены жители Средиземноморских стран, африканского и латиноамериканских континентов. По этой причине существует предположение, что болезнь выступает своеобразным защитным барьером организма против малярийного плазмодия.
Что представляет собой бета-талассемия

Красный пигмент крови гемоглобин (Hb) выражается в виде основных составляющих красных кровяных клеток – эритроцитов, которые играют весомую роль в насыщении кислородом тканей через легкие и устранении углекислого газа из тканей. Кроме того, элемент принимает участие в выводе продуктов обмена из организма.
Молекулярная структура гемоглобина достаточно тесно связана с его биологическим предназначением, ведь непосредственно сложное строение молекулы Hb способствует выполнению физиологических функций, заключающихся в транспортировке кислорода и питании клеток. Несомненно, эритроциты, приносящие нездоровый гемоглобин утрачивают способность качественно выполнять необходимую работу, что провоцирует развитие гипохромной анемии.
Гемоглобин в крови человека выражается в форме сложного белка, состоящего из 4 гемсодержащих субъедниц, включающих двухвалентное железо и несущих ответственность за способность насыщение кислородом крови. Глобин – белок, включенный в молекулярный состав Hb тесно связан с гемом. Глобин состоит из пары альфа- и бета- цепей, включающих в себя остатки аминокислоты (α-цепь – в количестве 141, β-цепь – 146).
Весомое число аномальных гемоглобинов образуется вследствие замены остатка аминокислоты в одной из цепочек глобина, что провоцирует точечную мутацию. Основная масса подмен приходится на β-цепи, по этой причине бета-талассемия охватывает широкую группу в области гемоглобинопатий, вызванных сбоями в процессе выработки белковой составляющей молекулы Hb. Совместно с этим α-цепь не обладает особым разнообразием, что сказывается на распространении альфа талассемии.
Почему происходит разрушение эритроцитов

Талассемия – это болезнь генетического происхождения, возникающая в случае наследования аномального гена от обоих родителей и его отсутствии в половых хромосомах. Прямым фактором возникновения патологии становятся разнообразные мутационные сбои в гене, кодирующем синтез одной било другой цепи Hb. В основу молекул патологии могут входить синтез дефектной аномальной мРНК, структурное изменение генетического материала, мутация и непродуктивная транскрипция регуляторных генов. Результатом таких патологий становится сокращение либо прекращение синтеза одной из полипептидных гемоглобиновых цепочек.
Таким образом, при бета-талассемии синтез β-цепей происходит в неполном объеме, а это вызывает формирование избыточных α-цепей, и наоборот. Излишне синтезированные полипептидные цепочки сохраняются в клетках эритроцидного типа, повреждая их. Процессу сопутствует разрушение эритрокариоцитов в костном мозге, гемолиз эритроцитов в периферической крови, уничтожением ретикулоцитов в селезенке. Помимо того, при бета-талассемии эритроциты накапливают фетальный Hb, не транспортирующий кислород в ткани, вследствие чего образуется тканевая гипоксия. Костномозговая гиперплазия провоцирует деформирование скелетных костей. Малокровие, гипоксия тканей и неэффективный эритропоэз в большей или меньшей степени вызывают нарушения в росте и развитии ребенка.
Разновидности бета-талассемии
Медициной признано несколько типов заболевания. Классификация основана на виде пораженной полипептидной цепи. По степени тяжести протекания патологию делят на:
- хроническую легкую;
- хроническую средне тяжелую;
- тяжелую.
Третья форма недуга может завершиться гибелью ребенка в период новорожденности. Согласно подходу и классификации медицинские работники различают талассемию следующих видов:
Кроме того, нередко патология делится на:
Обнаружить болезнь и его вид можно во время внутриутробного развития плода посредством специальных средств диагностики. Гетерозиготная форма β-талассемии протекает с незначительными симптомами либо с их полным отсутствием. Если терапия проведена своевременно, проявления могут исчезнуть и долгий период не тревожить больного.
Гомозиготная бета-талассемия становится прямым показателем к прерыванию беременности. Чаще всего гибель плода наступает еще до рождения или через некоторое время после него (1-2 года). В таких случаях объем фатального Hb в красных пигментах серьезно превосходит объем нормального.
Малая форма бета-талассемии
Образованию патологии способствуют процессы мутации в хромосоме 11й пары. Уровень гемоглобина пациентов в таком состоянии немного снижен. Гематологическая картина включает в себя:
- основную массу кровяных телец занимают микроциты;
- низкое содержание Hb в эритроцитах;
- анизопойкилоцитоз;
- небольшое расширение эритроцидного ростка в костном мозге;
- незначительное увеличение объема селезенки.
В некоторых случаях малая форма заболевания протекает без явных физических изменений. У человека отсутствуют жалобы, хотя у него могут диагностировать анемию легкого типа и пониженное объем эритроцитов. Геном включает в себя один здоровый и один частично или полностью дефективный. Биохимичесские исследования крови больного отражают высокую степень железа, лактатдегидрогеназы, фетального Gb.
Клиническая картина гетерозиготной талассемии может долгое время не проявляться или быть схожей с другими болезнями, например:
- недомогание;
- болевые ощущения в голове;
- головокружение.
При врачебном осмотре возможно обращение внимания на:
- бледно-желтый цвет кожи больного;
- увеличение объема печени и селезенки.
У детей с подобной патологией повышается восприимчивость к инфекциям, снижается иммунитет, по этой причине заболевания протекают тяжело.
У взрослых женщин беременность сопровождается развитием водянки плода. Нарушение несовместимо с жизнью малыша, при нормальных родах способствует образованию серьезных неврологических и психических патологий.
Минимальной бета-талассемией зовется синдром Сильвестрони-Бьянко. Подобный вид болезни проходит почти без проявлений и выявляется случайно в семьях, где встречается талассемия.
Большая бета-талассемия
Такая форма патологии (ее еще называют анемией Кули) несет самую большую опасность для больного. Это тяжело проходящая анемия, которая развивается очень стремительно. Симптомы присутствия аномального процесса появляются буквально с первых дней, достигают пика по прошествии 6 месяцев от рождения. Больной ребенок отличается бледностью кожных покровов с желтушным оттенком.
Кроме того, отмечаются характерные изменения на уровне физиологии в строении черепа, лица, недолжным образом образуется костная ткань, этому сопутствует замедление развития.
К первичным признакам относится рост объемов внутренних органов: печени и селезенки. Организм часто поражается заболеваниями инфекционного и воспалительного характера, вероятно формирование геморрагического синдрома. Нарушения в функционировании эндокринной системы возможны в следствие того, что дети не достигают периода половой зрелости.
Если пациенту, пораженному анемией Кули, своевременно не переливать кровь, возможно образование гипертрофии эритрпоэтических тканей. При регулярном проведении гемотрансфузии прогнозы не очень хорошие. Обычно средняя продолжительность жизни пациентов с легкой формой большой β-талассемии не превышает 20 лет, в исключительных случаях – 30 лет.
Диагностирование заболевания

Медицина предусматривает полный комплекс принципов диагностики, предназначенных для обнаружения у человека бета-талассемии. Выявить недуг можно даже при отсутствии клинической картины. Основой для установления диагноза служат:
- Присутствие особых патологий, разрушающих эритроциты.
- Нарушение обычного строения эритроцитов.
- Наследственное происхождение патологии.
При диагностике доктору требуются сведения о состоянии здоровья родителей больного для обнаружения вероятности генетически спровоцированных мутаций. Для этого также выполняется исследование крови на ДНК. Главным лабораторным способом установления диагноза талассемия становятся биохимические показатели крови, а именно:
- снижение показателей Hb до 50 г/л при гомозитном виде и до 110 г/л при гетерозиготном виде недуга;
- цветовой показатель не достигает 0,5;
- рост предшественников эритроцитов составляет менее 4%.
Тесты биохимии отражают сбои в обмене железа, как и в случае гемолитической анемии:
- повышенная степень свободного элемента билирубина;
- высокое содержание железа в сыворотке;
- пониженная возможность к связыванию железа.
Кроме лабораторных анализов крови проводится ряд исследований другого типа. Пункция костного мозга помогает выявить объем кроветворного ростка, краниограмма позволяет определить игольчатый периостоз. Ультразвуковое исследование брюшной полости способствует фиксированию измененных объемов печени и селезенки. При наличии подозрений на бета-талассемию докторам необходимо исключить различные типы анемий.
Терапия заболевания

Лечение бета-талассемии проводится согласно степени сложности эритропоэза, уровню поражения генов патологическими процессами. На сегодняшний день применяются следующие способы:
- питание пациента направлено на сокращение всасывания железа кишечником, рекомендации включают употребление орехов, сои, какао и чайных напитков;
- ежедневное удаление избытков железа посредством хелатов;
- к симптоматическим лекарствам относятся медикаменты гепатопротекторных функций – крупные дозы аскорбиновой кислоты устраняют излишки железа из организма;
- в случае резкого обострения назначаются большие дозы глюкокорткоидов;
- проведение спленэктомии (удаление селезенки) выполняется в случае увеличения органа у детей старше 5 лет. Самый подходящий возраст ребенка для процедуры 8-10 лет;
- при необходимости трансплантации ищется донор, соответствующий всем параметрам.
Талассемия любой формы нуждается в использовании медикаментов с фолиевой кислотой и витаминами группы В.
Лечение посредством гемотрансфузии
Больные с диагностированной тяжелой формой бета-талассемии нуждаются в регулярных процедурах переливания кровяной жидкости. Гемотрансфузия выполняется с первых недель жизни ребенка после появления на свет. Затем процедура повторяется с определенной регулярностью, что провоцирует развитие трансфузионной зависимости у больного. Переливание крови – единственный в своем роде процесс, который позволяет хотя бы на короткий период увеличить уровень Hb в крови. Рекомендации к проведению гемотрансфузии включают показатели гемоглобина на уровне 95-100 г/л.
Лечение народными рецептами

Безусловно, никакими растительными средствами побороть бета-талассемию нельзя, однако, некоторые способы позволяют облегчить проявление недуга и улучшить самочувствие больного, например:
- росту числа красных кровяных телец способствует прием свежевыжатого сока свеклы с ложкой натурального меда, по стакану трижды в день;
- смесь в одинаковых пропорциях свежих соков редьки, свеклы и моркови пьют трижды в день по 2 ст. л.;
- укрепляет состояние пораженных талассемией людей сок подорожника, который принимается в течение дня по 1 ст. л. перед каждым приемом пищи.
Требуется обязательное соблюдение диеты. Больным категорически запрещено употребление спиртного, жирного, жаренного и блюд быстрого приготовления.
Особую ценность для таких людей несут продукты, понижающие всасываемость железа. Для предотвращения деформации печени необходим прием гепатопротекторов.
Прогноз заболевания
Гемотрансфузия массы эритроцитов совместно с медикаментозным лечением обеспечивают улучшение качества жизни пациента. Несмотря на это, подобная терапия полностью диагноз не устраняет, потому как обладает поддерживающим предназначением. Жизнь людей, пораженных тяжелым видом талассемии, обычно не превышает более 7 лет при условии регулярного прохождения процедур гемотрансфузии. Болезненное деформирование внутренних органов отличается прогрессированием, что приносит ребенку страдания.
Профилактические меры

Профилактика бета-талассемии основана на предотвращении зачатия и появления на свет больных детей. Выполняется это несложно посредством простого исследования крови и процесса скрининга. Носителей заболевания предостерегают об угрозе связи с партнерами, пораженными таким же диагнозом.
Для снижения опасности рождения больного ребенка у пары с талассемией берется кровь на исследования различного рода. Для будущих мам, находящихся в положении, проводится полная дородовая диагностика, обследования генетического характера. Исследования ДНК и тесты ворсинок позволяют с начала беременности выявить гемоглобинопатию.
Чтобы оградить своих детей от любых заболеваний будущей мамочке необходимо всегда следить за своим здоровьем, а в случае наличия бета-талассемии, важное значение будет играть тщательный подход к выбору партнера.
На сегодняшний день заболевание считается неизлечимым. Новорожденный с легким типом бета-талассемии может прожить полноценную жизнь без серьезных последствий. Однако гарантировать непосредственно такую форму недуга современные ученые возможности не имеют. В особенности задуматься о деторождении нужно парам с гомозиготной наследственностью.







